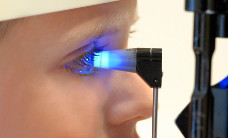
Retinoblastom (Kurzinformation)
Das Retinoblastom ist eine Krebserkrankung des Auges. In diesem Text erhalten Sie Informationen zu Krankheitsbild, Häufigkeit, möglichen Ursachen und Symptomen sowie zu Diagnose, Behandlung und Prognose der Erkrankung.
Autor: Maria Yiallouros, erstellt am: 04.04.2016, Freigabe: Dr. med. Christine Jurklies; PD Dr. med. Petra Temming, Zuletzt geändert: 12.06.2020 doi:10.1591/poh.retino-patinfo.kurz.1.20120611
Inhaltsverzeichnis
Krankheitsbild
Das Retinoblastom ist eine seltene Krebserkrankung des Auges. Es entsteht in der Netzhaut (Retina) und kommt fast ausschließlich im Kindesalter vor. Man unterscheidet eine erbliche und eine nicht-erbliche Form der Erkrankung. Im ersten Fall besteht eine Veranlagung für die Entwicklung dieser Krebsart, im zweiten Fall entsteht der Krebs spontan, das heißt durch eine neu aufgetretene Veränderung einer Netzhautzelle.
Ein Retinoblastom kann ein oder beide Augen betreffen. Meist erkrankt nur ein Auge (einseitiges oder unilaterales Retinoblastom); bei etwa einem Drittel der Kinder befällt die Krankheit beide Augen (beidseitiges oder bilaterales Retinoblastom). Ist Letzteres der Fall, so ist dies so gut wie immer ein Hinweis auf ein erbliches Retinoblastom. Einseitige Retinoblastome hingegen sind meist nicht erblich. Die Tumoren können sich entweder nur an einer Stelle im Auge (unifokal) oder an mehreren Stellen (multifokal) bilden.
Retinoblastome wachsen in der Regel schnell. Sie können sich innerhalb des Augapfels und, ausgehend von dort, auch in die Augenhöhle und entlang des Sehnervs in das Zentralnervensystem (ZNS) ausbreiten, in fortgeschrittenen Fällen auch über den Blut- und/oder Lymphweg in andere Organe. Wenn die Erkrankung unbehandelt bleibt, verläuft sie fast immer tödlich. Nur in seltenen Fällen (1-2 %) bildet sich der Tumor von selbst zurück; man spricht dann von einer spontanen Regression.
Häufigkeit
Das Retinoblastom ist der häufigste im Auge auftretende (intraokulare) bösartige Tumor im Kindesalter. In Deutschland erkranken pro Jahr etwa 40 Kinder unter 15 Jahren neu an dieser Krebsform. Das bedeutet, dass auf etwa 18.000 lebend geborene Kinder ein Kind mit einem Retinoblastom kommt. Insgesamt gesehen sind Retinoblastome allerdings selten: Sie machen nach Angaben des Deutschen Kinderkrebsregisters (Mainz) etwa 2 % aller bösartigen Erkrankungen im Kindes- und Jugendalter aus.
Das Retinoblastom tritt in der Regel bei Säuglingen und Kleinkindern auf, das heißt, es entsteht praktisch immer vor dem fünften Lebensjahr. Etwa 80 % der erkrankten Kinder sind jünger als vier Jahre. Jenseits des sechsten Lebensjahres entwickeln sich Retinoblastome extrem selten.
Ursachen
Ursache für die Entstehung eines Retinoblastoms sind zwei genetische Veränderungen (Mutationen) in den Vorläuferzellen der Netzhaut, den so genannten Retinoblasten. Solche Veränderungen können spontan in einzelnen Netzhautzellen auftreten. Sie können aber auch in den Keimzellen (und somit auch in allen Körperzellen) vorhanden sein und sind dann vererbbar.
Bei der Mehrheit der Patienten – etwa 60 % – handelt es sich um eine nicht-erbliche Form des Retinoblastoms, das heißt, die Mutationen entstehen isoliert neu (sporadisch) und befinden sich ausschließlich in den Tumorzellen. Etwa 40 % der Retinoblastome sind hingegen erblich. In ungefähr einem Viertel dieser Fälle – also bei insgesamt 10 - 15 % aller Patienten – sind bereits weitere Erkrankungen in der Familie bekannt (man spricht dann auch von einem familiären Retinoblastom); bei allen anderen erblichen Retinoblastomen entsteht die Krankheit neu.
Unabhängig davon, ob es sich um ein erbliches oder ein nicht-erbliches Retinoblastom handelt, stets liegen die genetischen Veränderungen im so genannten Retinoblastomgen, das sich auf Chromosom 13 befindet. Da jedes Chromosom doppelt vorhanden ist, gibt es auch zwei Retinoblastomgen-Allele in jeder Zelle. Nur wenn beide Allele verändert sind, kann sich ein Tumor entwickeln. Einzelheiten zur genetischen Einteilung der Retinoblastome und der Häufigkeit ihres Auftretens sowie zu ihrer Entstehung finden Sie in unserem Text "Vererbung / Genetik des Retinoblastoms".
Krankheitszeichen
Sehr kleine Retinoblastome bereiten in der Regel keine Beschwerden; die Erkrankung verläuft oft lange Zeit ohne Krankheitszeichen (Symptome). Beschwerden treten meist erst dann auf, wenn der Tumor größer ist oder in andere Teile des Auges einwächst. Dann kann es zur Beeinträchtigung des Sehvermögens oder gar zur Erblindung kommen. Durch die unterschiedliche Sehschärfeleistung der beiden Augen kann ein Schielen auftreten. Dies ist bei etwa 25 bis 30 % der Patienten der Fall.
Das häufigste Erstsymptom bei über zwei Drittel der erkrankten Kinder ist allerdings das weiße Aufleuchten der Pupille (Leukokorie) bei bestimmten Lichtverhältnissen, zum Beispiel beim Fotografieren – im Gegensatz zu einer rot aufleuchtenden oder einer schwarzen Pupille beim gesunden Auge. Diese weiße Pupille, auch als Katzenauge bezeichnet, ist ein Hinweis auf ein ausgedehntes Tumorwachstum hinter der Linse. Seltener fallen Kinder durch ein schmerzendes, gerötetes oder geschwollenes Auge auf, das auf einem erhöhten Augeninnendruck beruht.
Auf folgende Warnzeichen sollten Sie bei Ihrem Kind achten:
- Eine weißlich-gelbe Färbung einer oder beider Pupillen (Leukokorie)
- Schielen oder Abnahme der Sehschärfe/Sehstörung
- Rötung oder Schwellung des Auges; Augenschmerzen
Das Auftreten eines oder mehrerer dieser Krankheitszeichen muss nicht bedeuten, dass ein Retinoblastom oder eine andere Tumorerkrankung vorliegt. Einige dieser Symptome können auch vergleichsweise harmlose Ursachen haben, die mit einem Tumor nichts zu tun haben. Dennoch ist es ratsam, so bald wie möglich einen Arzt zu konsultieren, um die Ursache zu klären. Liegt tatsächlich ein Retinoblastom (oder eine andere bösartige Erkrankung) vor, so ist eine rechtzeitige Diagnose die beste Voraussetzung für eine erfolgreiche Behandlung der Krankheit.
Kinder aus Familien mit erblich bedingt erhöhtem Krankheitsrisiko müssen – auch ohne Vorliegen von Beschwerden oder Symptomen – regelmäßig augenärztlich untersucht werden, damit ein Retinoblastom im Frühstadium erkannt wird und entsprechende Behandlungsmaßnahmen rechtzeitig eingeleitet werden können.
Diagnose
Findet der (Kinder-)Arzt durch Krankheitsgeschichte (Anamnese) und körperliche Untersuchung Hinweise auf ein Retinoblastom, wird er den Patienten in ein Krankenhaus überweisen, das auf diese Form der Krebserkrankung spezialisiert ist (augenärztliche oder kinderonkologische Behandlungseinrichtung). Denn bei Verdacht auf ein Retinoblastom sind verschiedene Untersuchungen notwendig, zunächst um die Diagnose zu sichern, dann aber auch um festzustellen, um welche Form des Retinoblastoms es sich handelt (erblich oder nicht-erblich) und wie weit sich die Erkrankung ausgebreitet hat.
Die wichtigste Untersuchung zum Nachweis eines Retinoblastoms ist die Augenspiegeluntersuchung (ophthalmoskopische Untersuchung). Dabei wird mit Hilfe von Augenspiegeln und einer starken Lichtquelle der Augenhintergrund betrachtet. Wird tatsächlich ein Retinoblastom festgestellt, so sind weitere Untersuchungen notwendig, um die genaue Tumorausbreitung zu erfassen. Zu den wichtigsten Untersuchungsmethoden gehören die Ultraschalluntersuchung (Sonographie) und die Magnetresonanztomographie (MRT). Zusätzlich wird eine kinderärztliche Untersuchung durchgeführt.
In seltenen Fällen, beispielsweise bei extrem fortgeschrittener Erkrankung und/oder vor einer Chemotherapie, können weitere Untersuchungen hinzukommen (zum Beispiel eine Röntgenuntersuchung des Brustkorbs, eine Untersuchung der Gehirn-Rückenmarks-Flüssigkeit (Lumbalpunktion), des Knochenmarks (Knochenmarkpunktion) und/oder der Knochen (Skelett-Szintigraphie).
Die Diagnostik beschränkt sich nicht nur auf das erkrankte Kind. Da eine erbliche Form des Retinoblastoms vorliegen kann, sind auch eine augenärztliche Untersuchung der Geschwister und der Eltern sowie eine molekulargenetische Analyse von Blutproben (Gentest) notwendig, um Vererbungsrisiken zu klären. Wenn alle notwendigen Untersuchungen abgeschlossen sind, kann das Behandlungsteam mit Ihnen gemeinsam entscheiden, welche Behandlungsmaßnahmen Ihrem Kind am besten helfen.
Behandlung
Zur Behandlung von Patienten mit einem Retinoblastom stehen als Behandlungsformen die Operation, die Bestrahlung (Brachytherapie oder perkutane Strahlentherapie), die Lasertherapie, die Kryotherapie und die Chemotherapie zur Verfügung.
Welche Verfahren angewandt werden, hängt in erster Linie davon ab, ob ein oder beide Augen vom Tumor betroffen sind, wie weit die Erkrankung zum Zeitpunkt der Diagnose fortgeschritten ist (Krankheitsstadium) und ob für ein oder beide Augen nach der Therapie noch Sehfähigkeit zu erwarten ist. Auch das Alter des Kindes wird bei der Behandlungsplanung berücksichtigt. Ziel jeder Therapie ist die vollständige Zerstörung beziehungsweise Entfernung des Tumors und damit die Heilung der Krebserkrankung. Der Erhalt des Lebens steht dabei grundsätzlich über dem Erhalt des Sehvermögens.
Prinzipiell sind zwei Behandlungsstrategien möglich:
- die operative Entfernung des Tumors durch Entfernen des Auges (Enukleation)
- eine Augapfel-erhaltende Therapie mittels Strahlen-, Laser-, Kryo- und/oder Chemotherapie
Eine Augapfel-erhaltende Therapie kommt in der Regel nur dann in Frage, wenn das Retinoblastom frühzeitig erkannt wird. Sie hat das Ziel, den Tumor zu inaktivieren und gleichzeitig das Sehvermögen zu erhalten, ohne dass dabei ein Lebensrisiko eingegangen wird. Ist die Krankheit bereits fortgeschritten, ist die Entfernung des Auges meist unumgänglich. Sind Tochtergeschwülste (Metastasen) vorhanden, wird zusätzlich zur Operation eine Chemotherapie und/oder Strahlentherapie durchgeführt.
Behandlung von Patienten mit einseitigem Retinoblastom
Liegt ein einseitiges Retinoblastom vor, so ist die Entfernung des erkrankten Auges (Enukleation) die häufigste und auch die sicherste Behandlungsmethode, da bei einem funktionstüchtigen Partnerauge die Risiken anderer Behandlungsstrategien vermieden werden können. Bei nicht-erblichen Retinoblastomen kann mit dieser Behandlung eine vollständige Heilung erreicht werden.
Wenn der Tumor noch klein ist, kann in manchen Fällen auch eine Augapfel-erhaltende Therapie in Frage kommen: Bei sehr kleinen Tumoren ist eine augenerhaltende Therapien mit Hilfe der Laserkoagulation, Kryotherapie oder einer lokalen Strahlentherapie, der so genannten Brachytherapie, möglich. Patienten mit größeren Tumoren können mit einer systemischen Chemotherapie oder einer intraarteriellen Chemotherapie behandelt werden (siehe unten). Die augenerhaltende Behandlung ist jedoch nur sinnvoll, wenn neben der Kontrolle des Tumors auch ein ausreichendes Sehvermögen erhalten bleibt.
Einseitige Retinoblastome werden allerdings in der Regel erst sehr spät, also in fortgeschrittenen Krankheitsstadien erkannt; das betroffene Auge ist dann häufig schon erblindet. Die Entfernung des Auges bedeutet in diesem Fall für das Kind keine veränderte Wahrnehmung oder schlechtere Orientierung.
Kann der Tumor durch die Operation vollständig entfernt werden, ist im Anschluss keine weitere Behandlung erforderlich. Zeigt die feingewebliche Untersuchung des entfernten Auges jedoch, dass der Tumor sehr ausgedehnt war oder die Organgrenze bereits überschritten hatte, so erfolgt nach der Enukleation eine Chemotherapie, um eventuell im Körper verbliebene Tumorzellen oder kleine Metastasen zu vernichten. Nur selten ist zusätzlich eine perkutane Strahlentherapie erforderlich.
Behandlung von Patienten mit beidseitigem Retinoblastom
Ist Ihr Kind an einem beidseitigen (bilateralen) Retinoblastom erkrankt, so sind die Ärzte bemüht, durch eine individuelle Kombination der zur Verfügung stehenden Therapieverfahren den Tumor vollständig unter Kontrolle zu bringen und dabei gleichzeitig die Sehfunktion an zumindest einem Auge zu erhalten.
Therapie der Wahl ist zunächst eine lokale Behandlung. Einzelne kleine Retinoblastome lassen sich mit lokalen Therapieformen (Laserkoagulation, Kryotherapie oder Brachytherapie) sicher zerstören. Allerdings ist oft eine wiederholte Anwendung erforderlich. Sind die Tumoren für eine solche Behandlung bereits zu groß, kann in einigen Fällen eine Chemotherapie mit dem Ziel der Tumorverkleinerung (Chemoreduktion) durchgeführt werden, um anschließend eine lokale Behandlung (also eine Laser-, Kryo- oder Brachytherapie) möglich zu machen.
Häufig ist in einem der beiden Augen die Erkrankung allerdings so weit fortgeschritten, dass der Erhalt des Augapfels nicht sinnvoll erscheint und daher eine Enukleation erfolgt. In Fällen, in denen für die Behandlung des besseren Auges eine Chemotherapie in Betracht gezogen wird, kann manchmal mit der Enukleation des stärker betroffenen Auges zunächst gewartet werden. Denn durch die Therapie kann es zu einer starken Tumorrückbildung kommen, so dass dann doch noch eine Augapfel-erhaltende Behandlung möglich wird. Wenn das schlechtere Auge jedoch bereits erblindet ist oder eine Infiltration des vorderen Augensegments oder des Sehnervs besteht, gibt es keine Alternative zur Enukleation.
Schreitet auch im zweiten, zunächst besseren Auge die Erkrankung fort (Tumorbefall des Sehnervs oder der Aderhaut; Glaskörperaussaat), so bleibt oft als einzige Augapfel-erhaltende Therapie die perkutane Bestrahlung. Diese erfolgt auch wieder nur unter der Voraussetzung, dass ein ausreichendes Sehvermögen erhalten bleibt. Ist dies nicht der Fall, so muss auch das zweite Auge entfernt werden, um das Leben des Kindes nicht zu gefährden.
Da das Risiko, einen Zweittumor zu entwickeln, nach einer Strahlentherapie deutlich erhöht ist, ist man heute bestrebt, wenn möglich auf eine perkutane Strahlentherapie zu verzichten, insbesondere im ersten Lebensjahr. Allerdings bleibt die Wirksamkeit der perkutanen Bestrahlung bei dem sehr strahlenempfindlichen Retinoblastom unbestritten.
Intra-arterielle Chemotherapie
Mit dem Ziel, die Notwendigkeit einer Augapfelentfernung oder einer perkutanen Strahlentherapie weiter zu verringern und, wenn möglich, auch die Nebenwirkungen einer intravenösen, also den gesamten Körper betreffenden (systemischen) Chemotherapie zu vermeiden oder zu reduzieren, werden seit einiger Zeit neue Behandlungsmethoden entwickelt und erprobt.
Bei der intra-arteriellen Chemotherapie wird ein Zytostatikum (zum Beispiel Melphalan) ganz gezielt über eine Augenarterie in das Auge gegeben wird. Es wird dazu ein Katheter in die Leistenarterie eingeführt und am Herzen vorbei bis den Bereich der Augenarterie des betroffenen Auges vorgeschoben. Das verabreichte Medikament verteilt sich von dort aus im nachgeordneten Gefäßsystem und damit auch in den Tumorgefäßen des Retinoblastoms.
Studien und Register
Da das Retinoblastom eine sehr seltene Krankheit ist (in Deutschland und Österreich erkranken jährlich circa 44 Kinder pro Jahr), existieren bisher nur wenige Daten, die als Grundlage für eine risikoadaptierte (also eine an das jeweilige Rückfallrisiko des Patienten angepasste) und evidenzbasierte (das heißt, eine statistisch gesicherte) Auswahl der im Einzelfall am besten geeigneten Therapieverfahren dienen könnten.
Anders als bei anderen bösartigen Krebserkrankungen, die im Kindes- und Jugendalter auftreten können, gibt es für Patienten mit einem Retinoblastom bislang keine kontrollierten, standardisierten Therapievorgaben, zum Beispiel im Rahmen einer Therapieoptimierungsstudie. Aus diesem Grund wurde Ende 2013 das klinische Register RB-Registry eröffnet. Es soll mehrere Jahre lang Daten zur Epidemiologie und zum Krankheitsverlauf des Retinoblastoms erfassen mit dem Ziel, die Kenntnisse über die Erkrankung und ihr Ansprechen auf verschiedene Therapien zu verbessern.
In das Retinoblastomregister können deutschland- und österreichweit alle Kinder und Jugendlichen unter 18 Jahren aufgenommen werden, bei denen erstmalig ein Retinoblastom und/oder eine RB1-Mutation in der Keimbahn diagnostiziert wurde und die noch nicht keine Vorbehandlung erhalten haben. Die Zentrale des Registers befindet sich an der Universitätskinderklinik Essen unter der Leitung von PD Dr. med. Petra Temming.
Für Patienten mit einem erblichen Retinoblastom gibt es darüber hinaus die Möglichkeit, an einer europäischen Studie teilzunehmen, die das Auftreten von Zweitumoren nach Retinoblastomerkrankung untersucht (Studie „Screening auf Zweittumoren bei Kindern mit erblichem Retinoblastom“). Im Rahmen dieser Screening-Studie wird einmal jährlich eine Magnetresonanztomographie (MRT) des Kopfes durchgeführt. Aufnahmekriterien für die Studie sind: erbliches Retinoblastom, eine vorausgegangene Bestrahlung und ein Alter zwischen 8 und 18 Jahren.
Prognose
Über 95 % der Kinder mit Retinoblastom können heute dank moderner Diagnose- und Behandlungsverfahren langfristig von ihrer Erkrankung geheilt werden. Kinder mit einem einseitigen Retinoblastom haben ein gesundes Auge ohne Beeinträchtigung des Sehvermögens und können ein ganz normales Leben führen. Auch bei der Mehrzahl der Kinder mit beidseitigem Retinoblastom bleibt mindestens ein Auge mit einer ausreichenden Restsehschärfe erhalten.
Die Prognose für den einzelnen Patienten hängt in besonderem Maße davon ab, wie weit die Krankheit zum Zeitpunkt der Diagnose fortgeschritten ist (Krankheitsstadium) und ob ein erbliches oder nicht-erbliches Retinoblastom vorliegt.
Retinoblastome, die zum Zeitpunkt der Diagnose auf das Auge – oder die Augen – beschränkt sind (intraokulares Retinoblastom), können besser behandelt werden als Erkrankungen, die in ihrem Wachstum weiter fortgeschritten sind; sie gehen daher prinzipiell mit einer günstigeren Prognose einher.
Patienten mit einem erblichen Retinoblastom haben eine insgesamt ungünstigere Gesamtprognose als Patienten mit der nicht-erblichen Form. Das hängt damit zusammen, dass bei der erblichen Erkrankung, unabhängig von der Behandlung, ein genetisch bedingt erhöhtes Risiko besteht, an einem zweiten bösartigen Tumor (zum Beispiel einem Weichteiltumor oder einem Osteosarkom) zu erkranken. Dieses Risiko nimmt weiter zu, wenn im Rahmen der Behandlung eine Bestrahlung des Augapfels erfolgt. Etwa 5 % der Kinder mit einem erblichen, zunächst einseitigen Retinoblastom entwickeln innerhalb von eineinhalb Jahren nach der Ersterkrankung ein Retinoblastom auf der Gegenseite.
Literatur 
- Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Anual Report 2018 (1980-2017). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2019 [URI: www.kinderkrebsregister.de]
- Temming P, Arendt M, Viehmann A, Eisele L, Le Guin CH, Schündeln MM, Biewald E, Astrahantseff K, Wieland R, Bornfeld N, Sauerwein W, Eggert A, Jöckel KH, Lohmann DR: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatric blood & cancer 2017, 64: 71 [PMID: 27567086]
- Temming P, Viehmann A, Arendt M, Eisele L, Spix C, Bornfeld N, Sauerwein W, Jöckel KH, Lohmann DR: Pediatric second primary malignancies after retinoblastoma treatment. Pediatric blood & cancer 2015, 62: 1799 [PMID: 25970657]
- Temming P, Eggert A, Bornfeld N, Sauerwein W, Göricke S, Lohmann DR: [Diagnosis and treatment of retinoblastoma: current strategies for effective tumour control and preservation of vision]. Klinische Monatsblatter fur Augenheilkunde 2013, 230: 232 [PMID: 23508752]
- Shields CL, Shields JA: Intra-arterial chemotherapy for retinoblastoma: the beginning of a long journey. Clinical & experimental ophthalmology 2010, 38: 638 [PMID: 20584015]
- Shields CL, Shields JA: Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Current opinion in ophthalmology 2010, 21: 203 [PMID: 20224400]
- Lohmann D: Die Genetik des Retinoblastoms. WIR Informationsschrift der Aktion für krebskranke Kinder e.V. (Bonn) 2007, 1: 31 [URI: www.kinderkrebsstiftung.de]
- Jurklies C: Das Retinoblastom - Diagnose und Therapie. WIR Informationsschrift der Aktion für krebskranke Kinder e.V. (Bonn) 2007, 1: 26 [URI: www.kinderkrebsstiftung.de]
- Wieland R, Havers W: Retinoblastome, in: Gadner H, Gaedicke G, Niemeyer CH, Ritter J: Pädiatrische Hämatologie und Onkologie. Springer Medizin Verlag 2006, 823 [ISBN: 3540037020]
 PDF-Datei der Patienteninformation zum Retinoblastom (286KB)
PDF-Datei der Patienteninformation zum Retinoblastom (286KB)
Autor: Maria Yiallouros, Dr. med. Christine Jurklies
Stand: 12.06.2020