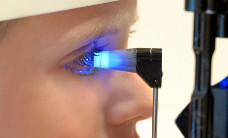
Retinoblastoma (brief information)
Retinoblastoma is a rare form of eye cancer that almost only occurs during childhood. The following pages provide information on possible causes and symptoms as well as on courses of the disease and its management, long-term follow-up and prognosis.
Author: Maria Yiallouros, erstellt am: 2016/04/04, Editor: Maria Yiallouros, Reviewer: Dr. med. Christine Jurklies; Dr. med. Petra Temming, English Translation: PD Dr. med. Gesche Tallen, Last modification: 2017/05/11 doi:10.1591/poh.retino-patinfo.kurz.1.20120611
Table of contents
General disease information
Retinoblastoma is a rare eye cancer arising from the retina, the inner layer of the eye, which is specialised in colour- and light perception. It almost only occurs during childhood. The development of retinoblastoma is initiated by genetic alterations (mutations) of the retinoblastoma (RB1) gene in immature retina cells (retinoblasts). There is a hereditary (congenital) and a non-hereditary form of the disease.
Retinoblastoma may affect one or both eyes. In the majority of patients, only one eye is affected (unilateral retinoblastoma); in approximately one third of the children, the tumour develops in both eyes (bilateral retinoblastoma). Bilateral disease is mostly seen with hereditary retinoblastoma, while most unilateral retinoblastomas are non-hereditary. Tumours may develop either at one site in the eye (unifocal) or at multiple sites (multifocal).
In general, retinoblastomas grow very fast. They can spread within the eyeball, into the orbit, and along the optic nerve into the central nervous system (CNS). In advanced stages, they may also spread into other organs via the blood and/or lymphatic system. If not treated appropriately, the outcome will be lethal. Only in very few patients (1 to 2 %), the tumour disappears without treatment (spontaneous regression).
Incidence
Retinoblastoma is the most common childhood cancer of the eye. About 40 children under 15 years of age are diagnosed with this type of malignancy in Germany each year. This means that there is one child with retinoblastoma among 18,000 children born alive. Overall, retinoblastoma is a rare disease. According to the German Childhood Cancer Registry (Deutsches Kinderkrebsregister) in Mainz, retinoblastomas account for approximately 2 % of all malignant diseases in children and adolescents.
Most retinoblastomas are diagnosed in infants and small children, i.e. before the fifth year of life. About 80 % of the patients are younger than four years old. Beyond the sixth year of life retinoblastomas are extremely rare.
Causes
The causes for a retinoblastoma to arise in the eye include certain genetic alterations (mutations) in the precursor cells of the retina, the so-called retinoblasts. Such alterations may occur spontaneously in single retinoblasts before or after the child's birth. Sometimes, however, the altered gene is transferred to the child by the germ cell of either parent or the mutation occurs at a very early stage of fetal development, thus causing the mutated gene to be present in all body cells, including all retinal cells and all germ cells (germline mutation). This means, the patient is born with a predisposition for the disease and can pass it on to his own offspring, too.
In the majority of patients (about 60 %), the altered gene is only found in the tumour itself. They have the non-hereditary (sporadic) type of retinoblastoma. About 40 % of retinoblastoma, though, are congenital (hereditary), which means, the affected children have a predisposition for the tumour to develop. Only about one quarter of the these children (10 to 15 %) are presenting with a family story of retinoblastoma, this means, other family members are known to have this type of cancer, too (familial retinoblastoma). In the remaining three quarters of patients with congenital retinoblastoma, the underlying genetical alterations are believed to have happenend spontaneously (see above).
Regardless of hereditary or non-hereditary, the mutations causing retinoblastoma are found in the retinoblastoma (RB1) gene, which is located on chromosome 13. Since in human cells chromosomes occur in pairs, there are also two RB-genes in each cell. If a retinoblast has at least one healthy RB1 gene, retinoblastoma won’t occur. Only when both RB genes are altered, the tumour will form.
Good to know: The RB1 gene is a so-called tumour suppressor gene. This means that it makes a protein (RB1 protein), which usually controls the growth of cells, thereby promoting them to mature into functional cells and preventing them from growing too fast. If the gene is mutated, it is unable to make a functional RB protein. Impaired RB1 protein lacks the capacity of helping retinoblasts to mature as well as to let them know when to stop dividing. As a result, they get out of control, divide rapidly, thereby forming a malignant tumour of the retina.
Symptoms
Very small retinoblastomas usually do not cause any complaints; the disease frequently progresses without any symptoms for a long period of time. Health problems usually occur with tumour growth into adjacent eye tissue. This can result in an impairment of vision or even loss of sight. Differences in visual acuity of the eyes lead to crossed eyes (strabismus) in about 25 to 30 % of patients.
The most common and typical sign, which is found in about two thirds of patients, is a white flashing of the pupil (white pupillary reflex, leukocoria) under certain light conditions, for example, after a flash photograph was taken – in contrast to the well-known red or black appearance of the pupils in healthy eyes. This condition, also referred to as “amaurotic cat's eye” is caused by expansive tumour growth behind the lens. Less often, patients also complain about irritated, swollen, red or painful eye as a result of increased eye pressure caused by the growing tumour.
Warning signs indicative of retinoblastoma may be:
- “amaurotic cat’s eye” (leukocoria)
- different colour of each eye
- crossed eyes (strabismus) along with vision problems
- irritated eye (redness of the white part of the eye, bulging of the eye, eye pain)
Occurrence of one or more of these symptoms, however, does not necessarily prove underlying retinoblastoma or any other malignancy. Some of these symptoms may be caused by relatively harmless health conditions that are not associated with cancer at all. Nevertheless, it is recommended to consider these symptoms as warning signs and therefore to consult a physician promptly upon their appearance. If retinoblastoma (or another malignant disease) is present, early diagnosis is the best precondition for a successful treatment of the disease.
Diagnosis
If a child’s medical history, presenting complaints and symptoms, as well as the physical, particularly the eye exam are suggestive of retinoblastoma, the patient should immediately be referred to a children’s cancer centre. There, diagnostics and treatment can be initiated and performed by specialists with the necessary expertise in childhood cancer.
Close collaboration between various specialists (such as paediatric oncologists, eye doctors, surgeons, radiologists, radiation oncologists, to name a few) as well as special tests are required to confirm the diagnosis and to determine the type of retinoblastoma (hereditary or non-hereditary) and how the disease has spread. Knowing these details is absolutely essential for optimal treatment planning and prognosis.
One essential initial diagnostic procedure for a young patient presenting with suspected retinoblastoma is the examination of the eye (fundoscopy). If a retinal tumour is seen, further tests are necessary to assess the disease characteristics such as size and spread. These tests include ultrasound as well as magnetic resonance imaging (MRI) of the eye.
Rarely, for example in patients with advanced stages of the disease and/or prior to chemotherapy, additional tests are performed, such as chest X-ray, spinal tab (lumbar puncture), bone marrow aspiration and biopsy, and/ or bone scan.
The diagnostic procedures are usually not limited to the affected child only. Children in families with a known mutation of the retinoblastoma (RB1) gene, or with a known family history of retinoblastoma, are recommended to get genetic testing for the RB1 gene (a special blood test which reveals the defect in the gene) shortly after birth. Based on the results, and even if the child does not present with any complaints or symptoms, the doctors will assess the risk of inheritance. They will also recommend how often the child should be seen by an eye doctor, in order to detect any tumour development early and to promptly initiate appropriate treatment.
After necessary diagnostics are completed, the doctors will discuss the treatment options with you.
Treatment
Children with retinoblastoma should be taken care of in a children’s cancer centre by specialists, because they provide the expertise necessary to manage this rare type of childhood malignancy.
Current treatment options for children with retinoblastoma include surgery, radiotherapy, laser therapy, cryotherapy, thermotherapy, and chemotherapy.
The appropriate type of treatment is individually assessed for each patient with retinoblastoma. Treatment planning is generally based on the child’s age, whether one or two eyes are affected by the tumour, how far the disease has already spread (disease stage), and how good the vision is at the time of diagnosis (disease stage).
The major aims of each treatment option are: to safe the patient’s life by getting rid of the malignancy, to – if possible – not remove the eyeball and to salvage as much eye sight as possible.
In general, the following two treatment strategies currently exist:
- surgical removal of the tumour by removal of the eyeball (enucleation)
- saving the eye by using radio-, laser-, cryo-, thermo, and/or chemotherapy
Saving the eyeball is only an option if retinoblastoma has been diagnosed at an early stage. The goal in this case is to kill the tumour cells while preserving as much vision as possible, but also making sure that the treatment is efficient enough so the patient won’t die from the disease. If retinoblastoma has been diagnosed at an advanced stage, enucleation is usually performed. If the tumour has spread into other parts of the body (metastasised), treatment may also include chemotherapy and/or radiation therapy in addition to surgery.
Treatment of patients with retinoblastoma in one eye (unilateral retinoblastoma)
For patients with an advanced tumour in one eye (unilateral retinoblastoma) and with already severe loss of vision at diagnosis, surgery – that is, the removal of the affected eye (enucleation) – is known to be the most efficient method of tumour control. Usually, an artificial eyeball (orbital implant), for example made of silicone, is implanted later. This implant is surgically connected with the eye’s muscles, so it moves as the original eyeball would.
The benefit of enucleation for patients with large unilateral tumours is that the treatment is limited to the sick eye only, leaving the healthy eye unaffected by any possible side-effects of treatment. However, the patient will completely lose vision in the treated eye. But considering that most patients with large tumours already present with severely reduced sight in that eye at diagnosis may justify this approach.
Important to know: Most patients with non-hereditary retinoblastoma can be completely cured by enucleation.
Patients who have been diagnosed with small retinoblastoma in one eye may sometimes benefit from an eye-preserving strategy, such as local radiation therapy (brachytherapy). This type of treatment, however, is only recommended for patients with remaining preservable eye sight and for whom sufficient anti-tumour effects can be guaranteed. The benefits of this approach in comparison to enucleation still need to be determined.
Patients with unilateral retinoblastoma, who underwent complete surgical removal of the tumour, usually do not require any additional treatment after surgery (adjuvant therapy). However, if histology of the removed eye reveals extended disease within the eye or even spread of malignant cells into adjacent tissue (like optic nerve), adjuvant chemotherapy is recommended in order to destroy both locally remaining tumour cells and potential small metastases in other parts of the body. Additional percutaneous radiotherapy is an option, but rarely required.
Treatment of patients with retinoblastoma in both eyes (bilateral retinoblastoma)
For patients with bilateral retinoblastoma, tumour control can be achieved by a combination of different treatments. The major goal is – in addition to destroy the cancer – to preserve vision in at least one eye.
Initial treatment of choice is local treatment, such as by means of laser coagulation, cryo-, thermo, or brachytherapy. In particular patients with small retinoblastomas can efficiently be treated by single or multiple applications of these local therapies. For patients presenting with larger-sized retinoblastoma, shrinking the tumours by chemotherapy (chemoreduction) prior to local treatment has proven to be a feasible approach.
For some children with bilateral retinoblastoma, who have been diagnosed with already advanced disease in one eye, such as spread into adjacent tissue (for example the optic nerve, the vitreous body, or outer eye layers), and/or complete vision loss, the removal of this eye (enucleation) is usually the treatment of choice.
For patients, who later develop progressive disease in the initially less affected eye, but still have remaining vision, percutaneous radiotherapy may be considered, often remaining the only possible therapy to preserve the eye. But if progressive disease has already caused blindness, enucleation of the second eye also is the only chance to safe the child’s life.
Considering that the risk of long-term side effects after chemotherapy seems to be lower than after percutaneous radiotherapy, the current trend is a radiation-sparing approach, especially for children younger than one year of age. However, since the efficacy of percutaneous radiation for the extremely radiosensitive retinoblastomas is proven, the up- and downsides of this treatment form always require thorough balancing on an individual basis.
New therapy approaches
The goal of new treatment methods for children with retinoblastoma is to reduce the necessity of enucleation or local radiotherapy, respectively, as well as to prevent as many side-effects of intravenous (systemic) chemotherapy as possible.
One of the most promising therapy approaches is the so-called intra-arterial chemotherapy, by which a cytostatic agent is given directly into the tumour’s blood vessels. This approach is still experimental, which means that it is currently only being applied in a few treatment centres and mostly to individual patients, whose disease did not respond to standard therapy. The preliminary results are promising, but long-term follow-up of these patients is necessary in order to assess the overall outcome, including long-term side-effects.
Trials and Registries
Since retinoblastoma is rare, so far only few data have been acquired that can be used as a basis for treatment strategies which both consider a patient’s individual risk of recurrent disease (risk-adapted therapy) and are statistically proven (evidence-based therapy).
In contrast to treatments of other childhood cancers, a standardised therapy plan (protocol) for the management of retinoblastoma still needs to be established, for example as a therapy optimising trial.
Therefore, the Retinoblastoma-Registry (RB-Registry) was opened for patients in Germany and Austria by the end of 2013. This clinical registry aims at gathering various data on retinoblastoma over several years, such as incidences and clinical courses of the different types of the disease as well as their response to different treatment forms, in order to fill the information gap mentioned above and, thus, to optimise current therapy and outcomes.
All children and adolescents under the age of 18 years who have been diagnosed with retinoblastoma or have a germline mutation of the retinoblastoma (RB1) gene and who have not yet received any retinoblastoma-specific treatment can be included in the RB-Registry. The headquarters of the RB-Registry are located in the Childhood Cancer Centre of the University of Essen, Germany. The registry is coordinated by the Dr. Petra Temming.
In addition to registration in the RB-Registry, patients with hereditary retinoblastoma, who are between 8 and 18 years old and who have received local radiotherapy, have the option to participate in a study investigating the risk of developing another, second cancer. The study is called „Screening for secondary malignancies in children with hereditary retinoblastoma“. The screening includes yearly magnetic resonance imaging (MRI) of the brain in order to detect the development of a brain tumour as early as possible.
Also, a study about local chemotherapy is underway.
Prognosis
Due to continuously optimised diagnostic procedures and treatment forms, more than 95 % of children with retinoblastoma can be cured of their disease today. Children with unilateral retinoblastoma still have one completely unaffected eye with normal vision. Their life quality may not differ from that of their healthy peers at all. Also, most patients with retinoblastoma in both eyes will keep a certain amount of visual acuity in at least one eye.
The individual prognosis primarily depends on the patient's stage of disease at diagnosis as well as on whether they have the hereditary or non-hereditary type of retinoblastoma.
Patients with tumour growth limited to the eye(s) only (intraocular retinoblastoma) usually have a bigger chance of successful treatment than children whose disease is further progressed at diagnosis.
The overall prognosis of children with hereditary retinoblastoma is generally less favourable compared to the outcome of patients with the non-hereditary form, because, regardless of the treatment, hereditary retinoblastoma is associated with a higher risk of developing a second malignant tumour somewhere else in the body (for example a soft tissue sarcoma or an osteosarcoma). This risk increases if radiotherapy forms part of the treatment.
About 5 % of children with hereditary, unilateral retinoblastoma develop the disease in the contralateral eye within one and a half years after diagnosis of the first tumour.
References 
- Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Jahresbericht / Annual Report 2016 (1980-2015). Institut für Medizinische Biometrie, Epidemiologie und Informatik (IMBEI), Universitätsmedizin der Johannes Gutenberg-Universität Mainz 2016 [URI: www.kinderkrebsregister.de]
- Houston SK, Murray TG, Wolfe SQ, Fernandes CE: Current update on retinoblastoma. International ophthalmology clinics 2011, 51: 77 [PMID: 21139478]
- Shields CL, Shields JA: Intra-arterial chemotherapy for retinoblastoma: the beginning of a long journey. Clinical & experimental ophthalmology 2010, 38: 638 [PMID: 20584015]
- Shields CL, Shields JA: Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Current opinion in ophthalmology 2010, 21: 203 [PMID: 20224400]
- Lohmann D: Die Genetik des Retinoblastoms. WIR Informationsschrift der Aktion für krebskranke Kinder e.V. (Bonn) 2007, 1: 31 [URI: www.kinderkrebsstiftung.de]
- Jurklies C: Das Retinoblastom - Diagnose und Therapie. WIR Informationsschrift der Aktion für krebskranke Kinder e.V. (Bonn) 2007, 1: 26 [URI: www.kinderkrebsstiftung.de]
- Wieland R, Havers W: Retinoblastome, in: Gadner H, Gaedicke G, Niemeyer CH, Ritter J: Pädiatrische Hämatologie und Onkologie. Springer Medizin Verlag 2006, 823 [ISBN: 3540037020]
- Gutjahr P: Retinoblastome, in: Gutjahr P (Hrsg.): Krebs bei Kindern und Jugendlichen. Deutscher Ärzte-Verlag Köln 5. Aufl. 2004, 499 [ISBN: 3769104285]
- Abramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology 1998, 105: 573-9; discussion 579 [PMID: 9544627]
- Wong FL, Boice JD Jr, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF Jr, Li FP: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA : the journal of the American Medical Association 1997, 278: 1262 [PMID: 9333268]
- Imhof SM, Moll AC, Hofman P, Mourits MP, Schipper J, Tan KE: Second primary tumours in hereditary- and nonhereditary retinoblastoma patients treated with megavoltage external beam irradiation. Documenta ophthalmologica. Advances in ophthalmology 1997, 93: 337 [PMID: 9665291]
- Eng C, Li FP, Abramson DH, Ellsworth RM, Wong FL, Goldman MB, Seddon J, Tarbell N, Boice JD Jr: Mortality from second tumors among long-term survivors of retinoblastoma. Journal of the National Cancer Institute 1993, 85: 1121 [PMID: 8320741]
- Draper GJ, Sanders BM, Kingston JE: Second primary neoplasms in patients with retinoblastoma. British journal of cancer 1986, 53: 661 [PMID: 3718823]
 PDF Brief Information on Retinoblastoma (256KB)
PDF Brief Information on Retinoblastoma (256KB)
Author: Maria Yiallouros, Dr. med. Christine Jurklies
04/04/2016