(© bilderzwerg - Fotolia.com)
Tumoren der Bauchspeicheldrüse (Kurzinformation)
Autor: Prof. Dr. med. Dominik T. Schneider, Dr. med. Ines Brecht, Redaktion: Maria Yiallouros, Zuletzt geändert: 20.02.2019
Inhaltsverzeichnis
In der Bauchspeicheldrüse können sich sowohl gutartige als auch bösartige Tumoren entwickeln. Im Kindes- und Jugendalter kommt dies allerdings sehr selten vor; weniger als zehn Kinder oder Jugendliche erkranken in Deutschland pro Jahr an einem solchen Tumor.
Aus diesem Grund gibt es nicht wie bei anderen Tumoren des Kindes- und Jugendalters Therapiestudien, in denen die beste Therapiestrategie an vielen Patienten geprüft worden ist. Die Experten des Registers für Seltene Tumorerkrankungen in der Pädiatrie (STEP) haben jedoch auf den folgenden Seiten die Erfahrungen zusammengefasst, die sie selbst sowie internationale Forschergruppen gewonnen haben.
Jedes Kind ist aber anders und es ist wichtig, die Therapie individuell auf jeden einzelnen Patienten auszurichten. Daher bietet STEP Ihnen eine kostenfreie Beratung an. Gerne kann sich Ihr behandelnder Arzt oder Sie selbst direkt an die Experten von STEP wenden: unter step@klinikumdo.de. Durch die Meldung an das STEP-Register können Sie außerdem helfen, die Erfahrung bei diesen seltenen Tumoren zu erweitern. Damit helfen Sie anderen Kindern, die in Zukunft an einer solchen Erkrankung leiden.
Im Folgenden informieren wir Sie über die im Kindes- und Jugendalter am häufigsten auftretenden Tumoren der Bauchspeicheldrüse.
Einführung zur Bauchspeicheldrüse
Die Bauchspeicheldrüse (Pankreas) liegt auf der Höhe der Nieren direkt unterhalb des Magens an der Rückwand der Bauchhöhle. Das längliche Organ wird in Pankreaskopf und Pankreasschwanz unterteilt. Der Pankreaskopf wird vom Zwölffingerdarm umschlossen.
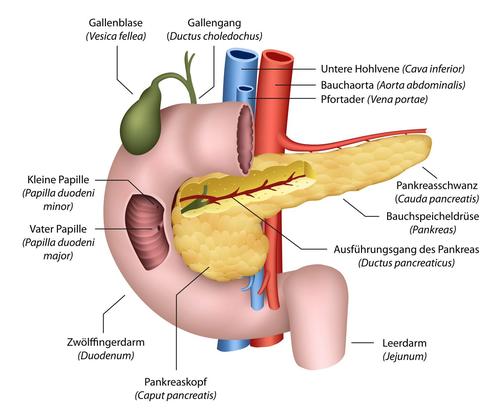
Die Bauchspeicheldrüse enthält sowohl exokrines als auch endokrines Drüsengewebe. Die exokrinen Drüsenzellen sind für die Bildung von Verdauungssäften (Enzymen) zuständig, die über den Bauchspeicheldrüsengang in den Zwölffingerdarm abgegeben werden. Das endokrine Gewebe besteht aus Zellverbänden, die verstreut in der Bauchspeicheldrüse liegen (so genannte Langerhans-Inseln). Sie bilden die Hormone Insulin und Glucagon, die den Blutzuckerspiegel steuern.
1. Das Pankreatoblastom
Das Pankreatoblastom ist ein embryonaler, bösartiger (maligner) Tumor der Bauchspeicheldrüse, der sehr selten vorkommt. Von 1972 bis 2012 wurde in der Fachliteratur von insgesamt gut 200 Fällen berichtet. Das mittlere Erkrankungsalter liegt bei fünf Jahren, allerdings fand sich der Tumor sowohl bei sehr kleinen Kindern als auch bei Jugendlichen und Erwachsenen.
Ursache: Wodurch entsteht ein Pankreatoblastom?
Bisher sind die Ursachen des Pankreatoblastoms nicht bekannt. Der Tumor wird allerdings gehäuft beobachtet, wenn Kinder an bestimmten genetisch bedingten Grunderkrankungen leiden (zum Beispiel einem Beckwith-Wiedemann-Syndrom). Dies legt die Vermutung nahe, dass Veränderungen im Bereich dieser Gene auch bei der Entstehung des Pankreatoblastoms eine Rolle spielen können.
Symptome: Welche Krankheitszeichen gibt es?
Da die Bauchspeicheldrüse im Bauchraum versteckt liegt, sind die Beschwerden oft unspezifisch und treten erst bei großen Tumoren auf. Häufige Symptome sind Bauchschmerzen und eine Umfangszunahme des Bauches beziehungsweise eine tastbare Struktur im Bauch. Es kann auch zu Gewichtsverlust und Abgeschlagenheit kommen, selten ist dagegen eine gelbliche Verfärbung der Haut. Der Stuhlgang kann entfärbt sein; er hat dann oft eine Farbe, die an Lehm oder Kreide erinnert.
Diagnose: Welche Untersuchungen werden durchgeführt?
In der Regel wird der (Kinder-)Arzt zunächst die Krankheitsgeschichte (Anamnese) erfragen und eine körperliche Untersuchung vornehmen. Anschließend erfolgt eine Routineblutentnahme. Diese beinhaltet ein Blutbild sowie die Untersuchung der Leberwerte und der Enzym der Bauchspeicheldrüse. Als spezieller Wert wird das so genannte Alpha-1-Fetoprotein (AFP) gemessen, der bei dieser Art von Tumor erhöht sein kann. Dieser Wert kann dann im Laufe der Therapie Informationen über das Ansprechen des Tumors auf die Behandlung geben. Man bezeichnet das AFP daher als Tumormarker.
Wichtig ist auch die optische Darstellung des Tumors, um Informationen über dessen Lage, das Wachstumsmuster und die Ausbreitung in benachbarte oder auch weiter entfernte Organe und Gewebe zu erhalten. Dies gelingt mit Hilfe verschiedener bildgebender Verfahren wie Ultraschall (Sonographie), Magnetresonanztomographie (MRT, Schichtaufnahme des Körpers mit Hilfe von Magnetfeldern) oder Computertomographie (CT, Schichtaufnahme des Körpers mittels Röntgenstrahlung).
Therapie: Wie erfolgt die Behandlung?
Der Tumor sollte operativ komplett entfernt werden. Ist dies nicht möglich, weil bereits lebenswichtige Organe oder Blutgefäße befallen sind, versucht man, vor der Operation den Tumor mit einer Chemotherapie zu verkleinern. Dabei kommen die Medikamente Cisplatin und Doxorubicin zum Einsatz. Die Therapie orientiert sich an den biologisch ähnlichen Lebertumoren des Kindesalters (Hepatoblastomen). Eine Strahlentherapie wird nur in seltenen Ausnahmefällen durchgeführt, wenn weder eine Chemotherapie noch eine Operation erfolgversprechend sind.
Wichtig: Die Tumoren verhalten sich aggressiv und erfordern eine optimale, an das jeweilige Rückfallrisiko der Erkrankung angepasste Behandlung. Lassen Sie sich daher bitte beraten.
Prognose: Wie sind die Heilungschancen?
Die Heilungschancen (Prognose) von Kindern und Jugendlichen mit Pankreatoblastom sind etwas besser als bei Erwachsenen mit bösartigen Pankreastumoren. Man geht davon aus, dass mehr als die Hälfte der Patienten langfristig geheilt werden kann.
Für die Prognose entscheidend ist, ob sich der Tumor schon in einem fortgeschrittenen Krankheitsstadium befindet, das heißt, ob sich Tochtergeschwülste (Metastasen) in Leber, Lunge, Knochen oder Gehirn gebildet haben. Denn je weiter sich der Tumor zum Zeitpunkt der Diagnose ausgebreitet hat, umso ungünstiger sind in der Regel die Heilungsaussichten. Besonders wichtig ist außerdem die bestmögliche Therapieplanung mit dem Ziel, eine komplette operative Tumorentfernung zu erreichen. Wichtig ist zudem eine sorgfältige Betreuung der Patienten während und nach Beendigung der Therapie durch speziell ausgebildete Kinderärzte und Kinderchirurgen.
2. Solide Pseudopapilläre Tumoren (SPT) des Pankreas
Solide Pseudopapilläre Tumoren (kurz: SPT) sind seltene Tumoren der Bauchspeicheldrüse. Seit 1959 wurde in der Literatur von mehr als 750 Erkrankungsfällen berichtet. Die meisten Fälle kommen im Erwachsenenalter vor. Auffällig ist die Häufung bei jungen Frauen. In gut 10 % aller Fälle sind Kinder und Jugendliche betroffen.
Ursache: Wodurch entsteht ein SPT?
Bisher gibt es keine Kenntnisse zum Entstehungsmechanismus von soliden pseudopapillären Tumoren. Ebenso sind keine Risikofaktoren bekannt.
Symptome: Welche Krankheitszeichen gibt es?
Das Symptombild kann stark variieren. In den meisten Fällen sind die Patienten komplett beschwerdefrei und der Tumor wird zufällig bei einer Routineuntersuchung entdeckt. Teilweise wird jedoch auch von Oberbauchschmerzen, Rückenschmerzen, Gelbfärbung der Haut, Übelkeit und Erbrechen oder einer tastbaren Struktur im Bauch berichtet.
Diagnose: Welche Untersuchungen werden durchgeführt?
In der Regel wird der (Kinder-)Arzt zunächst die Krankheitsgeschichte (Anamnese) erfragen und eine körperliche Untersuchung vornehmen. Anschließend erfolgt eine Routineblutentnahme. Diese beinhaltet ein Blutbild sowie die Untersuchung der Leberwerte und der Enzyme der Bauschspeicheldrüse. Anders als beim Pankreatoblastom (siehe oben) gibt es bei soliden pseudopapillären Tumoren keinen speziellen Laborwert (Tumormarker), der einen Hinweis auf diesen Tumor geben könnte. Es sollte jedoch eine Messung des Alpha-1-Fetoproteins im Blut erfolgen (AFP), um ein bösartiges Pankreatoblastom auszuschließen.
Wichtig ist auch die optische Darstellung des Tumors, um Informationen über dessen Lage, das Wachstumsmuster und die Ausbreitung in benachbarte oder auch weiter entfernte Organe und Gewebe zu erhalten. Dies gelingt mit Hilfe verschiedener bildgebender Verfahren wie Ultraschall (Sonographie), Magnetresonanztomographie (MRT, Schichtaufnahme des Körpers mit Hilfe von Magnetfeldern) oder Computertomographie (CT, Schichtaufnahme des Körpers mittels Röntgenstrahlung).
Therapie: Wie erfolgt die Behandlung?
Das Ziel der Therapie ist die vollständige operative Entfernung des Tumorgewebes. Gerade bei Kindern gelingt dies oft sehr gut. Ist eine vollständige Tumorentfernung nicht möglich, profitieren die Kinder und Jugendlichen auch von einer teilweisen Entfernung. Tochtergeschwülste (Metastasen) sind selten und sollten ebenfalls operativ entfernt werden. Chemo- und Strahlentherapie spielen in der Therapie keine Rolle oder werden allenfalls nur in sehr seltenen Fällen eingesetzt. Lassen Sie sich bei einem komplizierten Fall bitte beraten.
Prognose: Wie sind die Heilungschancen?
Patienten mit einem soliden pseudopapillären Tumor haben in der Regel eine sehr gute Prognose. 97% der Betroffenen leben nach fünf Jahren noch. Bei 15 % der Patienten kommt es innerhalb von etwa zehn Jahren zur Entwicklung von Metastasen im Bereich der Leber, des Bauchfells und, seltener, der Lymphknoten. Wichtig ist daher, dass die Patienten auch nach Ende der Therapie durch speziell ausgebildete Kinderärzte weiter betreut werden.
3. Andere Pankreastumoren bei Kindern und Jugendlichen
Aus den Hormon-bildenden Zellen der Bauchspeicheldrüse können sich so genannte neuroendokrine Tumoren entwickeln. Diese finden sich im Kindes- und Jugendalter gehäuft bei Patienten, die an einem multiplen endrokrinen Neoplasie-Syndrom (MEN-Syndrom) leiden. Sie sind in diesem Fall bedingt durch eine angeborene Mutation eines Krebsgenes.
Neuroendokrine Tumoren können gutartig oder bösartig sein; im letzteren Fall handelt es sich um Karzinome, die zum Teil einen aggressiven Verlauf nehmen und Metastasen ausbilden. Kinder und Jugendliche mit diesen Tumoren werden in Deutschland im Rahmen des GPOH-MET-Registers erfasst, das auch andere bösartige endokrine Tumoren im Kindes- und Jugendalter berücksichtigt. Dort werden Sie auch bezüglich der besten Behandlung beraten.
In der Bauchspeicheldrüse gibt es darüber hinaus bei Kindern und Jugendlichen sehr selten andere Karzinome, zum Beispiel das Azinuszellkarzinom oder das Adenokarzinom. Bei diesen Tumoren orientiert sich die Behandlung im Wesentlichen an den Empfehlungen für erwachsene Patienten. Für die Prognose entscheidend ist auch in diesen Fällen eine komplette operative Tumorentfernung. Bitte lassen Sie sich beraten.
Literatur 
- Schneider DT, Brecht IB: Das STEP-Register für seltene Tumorerkrankungen in der Pädiatrie - It's networking – or not working. WiR - die Zeitschrift der Deutschen Leukämie-Forschungshilfe e.V. und der Deutschen Kinderkrebsstiftung 3/2018 [URI: www.kinderkrebsstiftung.de]
- Schneider DT, Brecht IB, Olson ThA, Ferrari A (Eds): Rare Tumors In Children and Adolescents. Series: Pediatric Oncology, Springer-Verlag 2012 [ISBN: 978-3-642-04196-9]
- Bisogno G, Ferrari A, Bien E, Brecht IB, Brennan B, Cecchetto G, Godzinski J, Orbach D, Reguerre Y, Stachowicz-Stencel T, Schneider DT: Rare Cancers in Children - The EXPeRT Initiative: A Report from the European Cooperative Study Group on Pediatric Rare Tumors. Klin Pädiatr 2012, 224: 416 [PMID: 23143769]
- Schneider DT, Brecht I: Ein Netzwerk für besonders seltene Tumoren. WIR 2010, 3/10, 18 [URI: www.kinderkrebsstiftung.de]
- Brecht IB, Graf N, Schweinitz D, Frühwald MC, Bielack SS, Schneider DT: Networking for children and adolescents with very rare tumors: foundation of the GPOH Pediatric Rare Tumor Group. Klinische Padiatrie 2009, 221: 181 [PMID: 19437371]