Myelodysplastisches Syndrom (MDS) - Kurzinformation
Autor: PD Dr, med Ayami Yoshimi, erstellt am: 26.07.2020, Redaktion: Julia Dobke, Freigabe: Prof. Dr. med. Charlotte Niemeyer, Dr. Erlacher, Zuletzt geändert: 20.01.2021
Inhaltsverzeichnis
Krankheitsbild
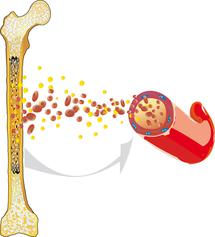
Das Myelodysplastische Syndrom (kurz: MDS) umfasst eine Gruppe von Erkrankungen, bei denen das Knochenmark nicht normal funktioniert und infolgedessen nicht ausreichend gesunde Blutzellen produziert werden. Alle Blutzellen, die im Blut vorkommen – dazu gehören rote Blutkörperchen (Erythrozyten), weiße Blutkörperchen (Leukozyten und Blutplättchen (Thrombozyten) – entstehen aus blutbildenden Zellen im Knochenmark, den so genannten Blutstammzellen). (Weiterführende Informationen hierzu erhalten Sie in unserer Patienteninformation zu Aufbau und Funktion von Knochenmark und Blut).
Bevor aus den Blutstammzellen im Knochenmark funktionsfähige Blutzellen entstehen können, müssen sie zahlreiche Reifungs- und Teilungsprozesse durchlaufen. Fachleute sprechen auch von Hämatopoese. Bei Patienten mit einem MDS sind diese Prozesse im Knochenmark gestört. Die Zellen reifen fehlerhaft aus, sie sehen auch unter dem Mikroskop anders aus als gesunde Zellen (Dysplasie). Diese fehlerhaft gebildeten Zellen gehen oft schon im Knochenmark zugrunde, so dass von dort aus nicht genügend Blutzellen ins Blut übertreten können. Die Folge davon ist, dass sich im Blut zu wenig gesunde rote und weiße Blutkörperchen sowie Blutplättchen befinden, was wiederum mit verschiedenen gesundheitlichen Problemen wie Blutarmut (Anämie), Fieber oder Blutungsneigung einhergeht.
Eine weitere Folge der gestörten Ausreifung von Blutstammzellen ist eine zunehmende Zahl unreifer Blutzellen (so genannter Blasten) im Knochenmark. Da die Blutzellen nicht vollständig ausreifen, können sie ihre vorgesehenen Funktionen nicht ausüben. Bei einem Teil der MDS-Patienten nimmt während des Krankheitsverlaufs die Zahl der Blasten im Knochenmark und im Blut so stark zu, dass das MDS nicht mehr von einer Leukämie unterschieden werden kann. Aus diesem Grund wurde die Erkrankung früher auch als „Präleukämie“ bezeichnet.
Häufigkeit
Das MDS ist die häufigste bösartige Erkrankung des Knochenmarks bei älteren Menschen, bei Kindern und Jugendlichen tritt es sehr selten auf. Bei Kindern unter 14 Jahren wird das MDS in weniger als 2 Fällen pro 1 Million Kinder im Jahr beobachtet. Das entspricht etwa 4 % aller Blutkrebserkrankungen im Kindes- und Jugendalter. In Deutschland werden circa 30 Kinder und Jugendliche pro Jahr mit einem MDS diagnostiziert. Jungen erkranken etwas häufiger als Mädchen.
Formen des MDS
Primäre und sekundäre MDS
Das MDS entsteht häufig ohne ersichtlichen Grund. Es wird dann als „primäres“ MDS bezeichnet. Etwa 75 % der Kinder mit einem MDS gehören in diese Gruppe. Es wird allerdings angenommen, dass auch bei einem primären MDS im Kindesalter angeborene genetische Veränderungen vorliegen können, die bisher noch nicht identifiziert wuden (siehe auch Abschnitt "Ursachen“).
Bei einem Teil der Patienten kann die Entstehung eines MDS mit bestimmten Auslösern in Verbindung gebracht werden; in diesen Fällen spricht man von einem „sekundären“ MDS. Manche Patienten erkranken zum Beispiel nach vorangegangener Behandlung mit einer Strahlentherapie oder Chemotherapie, die aufgrund einer anderen, meist bösartigen Erkrankung durchgeführt wurde. Andere Patienten leiden vor der Diagnose eines MDS bereits an einer angeborenen Erkrankung, die mit einer Störung der Knochenmarkfunktion einhergeht. Dazu gehören zum Beispiel die Fanconi-Anämie, die Dyskeratosis congenita, das Shwachman-Diamond-Syndrom, die Blackfan-Diamond-Anämie (DBA) oder eine schwere angeborene Neutropenie. Auch eine erworbene aplastische Anämie (SAA) kann der Diagnose eine MDS vorausgehen.
Untertypen des primären MDS im Kindesalter nach WHO-Klassifikation
Das primäre MDS bei Kindern wird nach der Klassifikation der Weltgesundheitsorganisation (englisch World Health Organization, WHO), der WHO-Klassifikation, in zwei Unterformen (Subtypen) eingeteilt. Diese Einteilung berücksichtigt hauptsächlich den Anteil der unreifen Zellen (Blasten) im Blut und im Knochenmark.
Refraktäre Zytopenie des Kindesalters (RCC)
Bei Patienten mit einer „refraktären Zytopenie des Kindesalters“ (RCC) ist der Blastenanteil mit unter 2 % im Blut und unter 5 % im Knochenmark im Vergleich zu gesunden Kindern nicht deutlich erhöht. Sehr häufig ist aber das Knochenmark bei Kindern mit RCC auffallend zellarm, das heißt, alle Zellen, die normalerweise im Knochenmark vorhanden sind (rote Blutzellen, weiße Blutzellen, Blutplättchen), liegen in reduzierter Zahl vor. Deswegen muss immer abgeklärt werden, ob es sich um andere angeborene Erkrankungen des Knochenmarks (Knochenmarkversagen) oder eine aplastische Anämie handelt, da auch bei diesen Erkrankungen ein zellarmes Knochenmark die Regel ist.
Myelodysplastisches Syndrom mit Blastenexzess (MDS-EB)
Bei MDS-Patienten kann der Anteil der Blasten in Blut und Knochenmark deutlich über 2 % beziehungsweise 5 % ansteigen. Dieser MDS-Typ wird als MDS-EB, also MDS mit Blastenexzess (englisch „excess of blasts“, EB), bezeichnet. Der Blastenanteil sollte allerdings 29 % im Knochenmark nicht übersteigen, denn in diesem Fall spricht man von einer Leukämie. Da bei der Unterscheidung von MDS und Leukämie aber auch andere Gesichtspunkte berücksichtigt werden, zum Beispiel die Geschwindigkeit, mit der die Blastenzahl zunimmt, ist die Unterscheidung (Differenzialdiagnose) zwischen MDS-EB und Leukämie manchmal schwierig. In Grenzfällen, das heißt, bei einer Blastenzahl zwischen 20 und 30 %, ist es sinnvoll, die Diagnostik (in Form einer Knochenmarkpunktion) nach zwei Wochen zu wiederholen, um die Diagnose zu sichern.
|
MDS-Subtyp |
Blut |
Knochenmark |
|---|---|---|
|
Refraktäre Zytopenie des Kindesalters (RCC) |
weniger als 2 % Blasten |
weniger als 5 % Blasten |
|
Myelodysplastisches Syndrom mit Blastenexzess (MDS-EB) |
2-29 % Blasten |
5-29 % Blasten |
Ursachen
Die genauen Ursachen für ein MDS bleiben meist ungeklärt; die Erkrankung ist aber – ebenso wie andere Krebsformen – weder ansteckend noch kann sie auf andere Menschen übertragen werden. In den meisten Fällen entsteht ein MDS bei zuvor gesunden Kindern und Jugendlichen ohne ersichtlichen Grund (so genanntes „primäres MDS“). Man geht aber insgesamt davon aus, dass die Betroffenen eine besondere Empfindlichkeit (Veranlagung) für die Entwicklung eines MDS beziehungsweise einer Leukämie aufweisen. Diese Veranlagung bezeichnet man auch als "Prädisposition".
Ursache der Prädisposition sind Genveränderungen (Mutationen), die auch in den Samen- oder Eizellen (also in der Keimbahn) vorliegen und dadurch vererbt werden können. Die Genveränderung liegt in diesem Fall auch in allen Körperzellen des Patienten vor. Die betroffenen Kinder und Jugendlichen haben diese Keimbahnmutation entweder von einem oder beiden Elternteilen vererbt bekommen, oder aber die Genveränderung des Erkrankten ist in der befruchteten Eizelle neu entstanden.
Es ist schon seit vielen Jahren bekannt, dass Genveränderungen, die angeborene Störungen der Blutbildung im Knochenmark verursachen können (wie die Fanconi-Anämie, die schwere angeborene Neutropenie, Dyskeratosis congenita oder Blackfan-Diamond-Anämie), auch mit einer Veranlagung für das Auftreten eines MDS einhergehen. In den letzten Jahren wurden weitere erbliche Erkrankungen entdeckt, die hier eine Rolle spielen. Dazu gehört die so genannte GATA2-Defizienz oder das SAMD9/SAMD9-Syndrom. Da diese angeborenen Erkrankungen in der Regel mit einem erhöhten Krebsrisiko einhergehen, werden sie auch als Krebsprädispositionssyndrome bezeichnet.
Bei älteren Menschen verläuft die Entwicklung eines MDS anders. In den meisten Fällen liegen bei Erwachsenen nicht angeborene Störungen, sondern über die Jahrzehnte erworbene Genveränderungen vor, die eine Zelle langsam zur MDS- oder Leukämiezelle werden lassen. Einige Patienten haben vor der Diagnose eines MDS eine Strahlentherapie oder Chemotherapie als Behandlung für eine andere Krebserkrankung erhalten. Das MDS ist dann die Zweiterkrankung (sekundäres MDS), die zumindest zum Teil durch die Behandlung der ersten Krebserkrankung ausgelöst wurde
Krankheitszeichen
Die Krankheitszeichen (Symptome), die bei einem Patienten mit MDS auftreten können, richten sich vor allem danach, wie stark der Mangel an (funktionstüchtigen) Blutzellen ausgeprägt ist, das heißt, nach dem Grad der (Zytopenie). Je nachdem, welche Zellen des Blutes betroffen sind, lassen sich folgende Formen der Zytopenie und damit einhergehend folgende Symptome unterscheiden:
Blutarmut: Mangel an roten Blutkörperchen (Anämie)
Die Aufgabe der roten Blutzellen (Erythrozyten) ist es, den Sauerstoff, der beim Einatmen über die Lunge aufgenommen wird, zu den verschiedenen Organen und Geweben des Körpers zu transportieren. Ein Mangel an roten Blutkörperchen (Anämie) führt zu Krankheitszeichen wie Blässe, Müdigkeit, Schwäche und Kopfschmerzen.
Abwehrschwäche: Mangel an weißen Blutzellen (Leukopenie/Neutropenie)
Weiße Blutkörperchen (Leukozyten) sind für die Abwehr von Krankheitserregern und somit für die Vermeidung von Infektionen verantwortlich. Es gibt verschiedene Arten von Leukozyten, zum Beispiel Lymphozyten und Granulozyten, die unterschiedliche Aufgaben in der Immunabwehr wahrnehmen. Durch den Mangel an funktionstüchtigen weißen Blutkörperchen, der so genannten Leukopenie, ist der Körper infektgefährdet. Bei Patienten mit einem MDS ist häufig in besonderem Maße die Zahl der Granulozyten vermindert (so genannte Granulozytopenie oder Neutropenie). Da Granulozyten für die Abwehr von Bakterien und Pilzen zuständig sind, treten bei MDS-Patienten insbesondere Infektionen mit Bakterien und Pilzen auf, die sich vor allem durch Fieber bemerkbar machen.
Blutungsneigung: Mangel an Blutplättchen (Thrombozytopenie)
Blutplättchen (Thrombozyten) spielen eine wichtige Rolle bei der Blutgerinnung. Sind sie vermindert (so genannte Thrombozytopenie), kommt es sowohl spontan als auch nach Verletzungen schneller zu Blutungen. Diese äußern sich zum Beispiel durch punktförmige Haut- oder Schleimhautblutungen (Petechien), blaue Flecken (Hämatome) und/oder Zahnfleischbluten, aber auch schwerwiegende Blutungen der inneren Organe oder des Gehirns kommen vor. Das Risiko für schwere Blutungen ist umso höher, je ausgeprägter die Thrombozytopenie, das heißt, je größer der Mangel an Thrombozyten ist.
Bei Patienten mit einem MDS mit Blastenexzess (MDS-EB) (siehe auch Abschnitt "Formen des MDS“) können schwer zu behandelnde Begleiterscheinungen auftreten, zum Beispiel eine Entzündung kleiner Blutgefäße oder das so genannte Sweet-Syndrom, das mit Fieber und roter Knötchenbildung auf der Haut einhergeht.
Diagnose
Findet der (Kinder-)Arzt durch Krankheitsgeschichte (Anamnese) und körperliche Untersuchung des Patienten Hinweise auf eine Bluterkrankung, wird er zunächst eine umfassende Blutuntersuchung vornehmen. Wenn sich, durch bestimmte Veränderungen im Blutbild, der Verdacht auf eine Blutkrankheit wie die MDS erhärtet, ist eine Entnahme von Knochenmark zur Sicherung der Diagnose notwendig. Zu diesem Zweck und für eventuell sich anschließende Untersuchungen wird der Arzt den Patienten in ein Krankenhaus überweisen, das auf Krebs- und Bluterkrankungen bei Kindern und Jugendlichen spezialisiert ist (Klinik für pädiatrische Onkologie/Hämatologie).
Untersuchung von Blut und Knochenmark
Da die Symptome bei MDS-Patienten nicht spezifisch auf ein MDS hinweisen und auch bei anderen Bluterkrankungen wie Leukämien auftreten, kann die Diagnose dieser Erkrankung nur durch eine umfassende Untersuchung des Blutes und des Knochenmarks gestellt werden.
Für die eindeutige Diagnosestellung des MDS wird Knochenmark benötigt, das aus dem Beckenknochen entnommen wird. Dafür sind eine Knochenmarkaspiration (Knochenmarkpunktion) und eine Knochenmarkstanzbiopsie notwendig. Beide Methoden ergänzen sich und werden im Rahmen einer gemeinsamen Untersuchung durchgeführt. Bei der Knochenmarkaspiration wird der Knochen mittels einer speziellen Hohlnadel punktiert und eine kleine Menge Knochenmarkblut angesaugt („aspiriert“). Bei der Knochenmarkstanzbiopsie wird ein kleines Stück des Beckenknochens (circa 1 mm Durchmesser) mittels einer Hohlnadel entnommen. Weitere, allgemeine Informationen zu Knochenmarkpunktion und -stanzbiopsie finden Sie hier.
Nach Gewinnung der Probe wird unter dem Mikroskop das Aussehen der blutbildenden Zellen im Knochenmark durch einen Spezialisten für Blut- und Krebserkrankungen beurteilt. Bei einem MDS sind verschiedene Veränderungen des Aussehens der Zellen (Dysplasie) möglich, die für die Diagnose entscheidend sind. Bei einem MDS-EB ist die Zahl der Blastenim Knochenmark erhöht (siehe „Formen: Welche unterschiedlichen Formen des MDS gibt es?“). Am Material aus dem Knochenmarkaspirat wird zusätzlich eine Untersuchung der Chromosomen (Zytogenetik) durchgeführt (siehe unten). Bei Verdacht auf das Vorliegen eines MDS sollten die Knochenmarkpunktion und die Knochenmarkstanzbiopsie nach ca. 14 Tagen wiederholt werden, um eine sichere Diagnosestellung zu gewährleisten.
Untersuchungen der Chromosomen (Zytogenetik)
Veränderungen der Chromosomen in Knochenmark und BlutZellen können bei mehr als der Hälfte der Patienten mit einem MDS mit Blastenexzess (MDS-EB) und bei etwa 30 % der Kinder mit einer refraktären Zytopenie (RCC) festgestellt werden. Der Nachweis dieser Veränderungen hilft bei der Bestätigung der Diagnose eines MDS. Die häufigste und eine typische Chromosomenveränderung bei Kindern mit MDS ist der Verlust eines Chromosoms 7. In diesen Fällen liegt das Chromosom 7 nur einmal anstatt zweimal in den Zellen vor (so genannte Monosomie 7).
Molekulargenetische Untersuchungen
Einige Kinder und Jugendliche mit einem MDS haben eine genetisch bedingte Empfindlichkeit (Prädisposition) für ein MDS, das heißt, bei ihnen liegt eine so genannte Keimbahnmutationen vor (siehe Kapitel „Ursache“). Für die Analyse dieser genetischen Veränderungen müssen neben Blut und/oder Knochenmark auch andere Körpergewebe wie zum Beispiel Haarwurzelzellen (Keimbahnmaterial) untersucht werden. Wird bei einem Patienten mit MDS eine Keimbahnmutation nachgewiesen, so muss im Falle einer geplanten Stammzelltransplantation (SZT) auch ein möglicherweise passender Familienspender auf diese Mutation hin untersucht werden. Nur wenn beim Familienmitglied die Mutation nicht nachweisbar ist, sollte es Stammzellen spenden.
Die Behandlung eines MDS richtet sich danach, welche Unterform der Erkrankung vorliegt (siehe Abschnitt “Formen des MDS“). Zur Behandlung stehen grundsätzlich folgende Therapiemaßnahmen zur Verfügung:
Therapie
- Transplantation von blutbildenden Stammzellen eines Spenders (allogene Stammzelltransplantation, SZT)
- immunsuppressive Therapie (IST)
- andere medikamentöse Therapien: Azacitidin, Chemotherapie
- unterstützende Therapien (Supportivtherapie): Transfusionen, Behandlung mit Antibiotika
Refraktäre Zytopenie des Kindesalter (RCC)
Die Patienten mit RCC werden entsprechend des Vorliegens von Chromosomenveränderungen in Blut- und Knochenmarkzellen, dem Ausmaß der Zellarmut im Blut (Zytopenie) und dem Zellgehalt (Zellularität) des Knochenmarks unterschiedlich behandelt.
Die RCC-Patienten, die in der Chromosomenanalyse von Knochenmarkzellen einen Verlust eines Chromosoms 7 (einer Monosomie 7 zeigen, haben einen ungünstigen Verlauf mit dem Risiko einer Blastenvermehrung innerhalb von 1 – 2 Jahren. Daher sollen diese Patienten so bald wie möglich mit einer allogenenStammzelltransplantation (SZT) behandelt werden (siehe auch „Therapie: Wie werden Kinder und Jugendliche mit einem MDS behandelt?“). Bei der Stammzelltransplantation werden dem Patienten zunächst hohe Dosen an Chemotherapie (Konditionierung) verabreichet, um im Knochenmark alle Zellen (gesund und krank) zu zerstören, das Knochenmark sozusagen zu leeren. Danach werden dem Empfänger gesunde Blutstammzellen aus dem Knochenmark oder dem peripheren Blut eines Spenders verabreicht. Mehr Informationen zur Stammzelltransplantation finden Sie hier.
Im Gegensatz dazu kann das Krankheitsbild bei RCC mit normaler Chromosomenanalyse über Jahre stabil sein. Wenn diese Patienten keine Bluttransfusionen benötigen und genügend Granulozyten (weiße Blutkörperchen, die Bakterien und Pilze abwehren) besitzen, wird das MDS zunächst nicht behandelt, sondern der Krankheitsverlauf ohne Therapie mit regelmäßigen Blutkontrollen und Untersuchungen beobachtet (sogenannte „watch and wait“-Strategie). Verschlechtern sich allerdings die Blutwerte mit der Zeit, kann im Verlauf der Erkrankung noch eine Therapie notwendig sein. Das ist dann in der Regel eine Stammzelltransplantation.
Im Gegensatz dazu werden Patienten, die schon nach der Diagnosestellung Bluttransfusionen brauchen oder sehr wenige Granulozyten im Blut haben, so bald wie möglich mit einer Stammzelltransplantation behandelt. Als Stammzellspender kommen gesunde Geschwister oder passende Fremdspender in Frage.
Ist kein passender Geschwisterspender verfügbar, kann auch eine immunsuppressive Therapie (IST) bei Patienten mit hypozellulärem Knochenmark die Therapie der Wahl sein. Diese immunsuppressive Behandlung ist ähnlich wie die Behandlung einer schweren aplastischen Anämie (SAA) und zieht sich über mehrere Monate oder Jahr hin.
Durch die IST kann bei etwa der Hälfte der Patienten ein ausreichender Anstieg der Blutzellenzahl erreicht werden, so dass in der Regel keine Bluttransfusionen mehr notwendig sind. Es ist auch möglich, dass nach zunächst erfolgreicher IST wieder eine Zytopenie, also ein Mangel an Blutzellen, entsteht (sogenanntes Rezidiv). In den Fällen mit einem Rezidiv oder bei Nicht-Ansprechen auf die IST-Therapie wird eine Stammzelltransplantation von einem gut passendem Fremdspender empfohlen.
Fortgeschrittenes MDS (MDS-EB)
Die Patienten mit einem MDS-EB (siehe “Formen: Welche unterschiedlichen Formen des MDS gibt es?“) haben ein sehr hohes Risiko, im weiteren Verlauf eine Leukämie zu entwickeln. Für ein MDS-EB ist eine frühzeitige Stammzelltransplantation in der Regel die Therapie der Wahl.
Als Stammzellspender kommen gesunde Geschwister oder passende Fremdspender in Frage. Bei höheren Blastenzahlen kann die Vorbehandlung mit einer Chemotherapie zur Blastenverminderung vor Stammzelltransplantation sinnvoll sein. Azacitidin (Vidaza®) ist eine Substanz, die in den Zellen wichtige Gene reaktiviert, die durch die Erkrankung fälschlicherweise abgeschaltet wurden (sogenannte epigenetische Therapie). Eine Therapie mit Azacitidin ist zwar nicht ausreichend für die Heilung eines MDS, aber sie kann bei Erwachsenen mit MDS die Überlebenszeit nachweislich verlängern. Bei Kindern mit MDS-EB kann eine Azacitidin-Therapie vor Stammzelltransplantation erfolgen, um die Blastenzahl effektiv zu reduzieren.
Sekundäres MDS
Patienten mit sekundärem MDS werden in der Regel wie Patienten mit einem primären MDS-EB behandelt. Diese Patienten sollen daher so bald wie möglich nach der Diagnosestellung von einem passenden Stammzellspender transplantiert werden.
Unterstützende (supportive) Therapie während der Behandlung
Für alle Patienten mit einem MDS sind unterstützende Therapiemaßnahmen (Supportivtherapien) sinnvoll und erforderlich. Diese zusätzlichen Therapien tragen dazu bei, krankheitsbedingte Symptome und behandlungsbedingte Nebenwirkungen zu behandeln oder diesen vorzubeugen. (siehe auch: https://www.kinderkrebsinfo.de/patienten/behandlung/behandlungsmethoden/supportivtherapie/index_ger.html)
Die meiste Patienten haben bei Diagnose eine Blutarmut (Anämie und / oder einen Mangel an Blutplättchen (Thrombozytopenie), die mit gesundheitlichen Problemen einhergehen (siehe „Krankheitszeichen: Welche Beschwerden haben Kinder und Jugendliche mit einem MDS?“).
Diese Probleme können durch Bluttransfusionen (Gabe von roten Blutzellen beziehungsweise Blutplättchen) behandelt werden. Allerdings wird bei wiederholten Transfusionen von roten Blutzellen dem Körper eine große Menge von Eisen zugeführt, das sich im Lauf der Zeit in Organen (vor allem der Leber und dem Herzen) ablagert und diese schädigen kann (sogenannte Eisenüberladung).
Bei Patienten mit einer Eisenüberladung muss daher eine Eisenentzugstherapie durchgeführt werden (siehe auch „https://www.kinderblutkrankheiten.de/content/erkrankungen/rote_blutzellen/sekundaere_eisenueberladung/).
Durch die Erkrankung MDS, aber auch durch deren Behandlung wie eine Stammzelltransplantation oder eine immunsuppressive Therapie, werden die Patienten in ihrer Immunabwehr geschwächt.
Die Patienten müssen daher so gut wie möglich vor Infektionen geschützt und im Falle des Auftretens von Infektionen schnellstmöglich behandelt werden. Fieber ist oft das erste Anzeichen für eine Infektion. Infektionen bei immungeschwächten Kindern sind stets als lebensbedrohlich anzusehen. Eltern oder Angehörige von Patienten, bei denen Fieber auftritt, sollten daher umgehend die behandelnde Klinik oder den betreuenden Arzt kontaktieren (auch nachts), damit die Kinder sofort mit Breitspektrum-Antibiotika behandelt werden können.
Bei Patienten mit einer Neutropenie (Mangel der Granulozyten) ist als unterstützende Therapie die vorbeugende Verabreichung von Antibiotika und Antimykotika (Antipilzmittel) angezeigt.
Prognose
Das MDS kann sehr unterschiedlich verlaufen. Es gibt Erkrankungen, die über einen langen Zeitraum stabil sind, andere schreiten rasch fort. Die Heilungsaussichten (Prognose) für MDS-Patienten wird durch die Art (Subtyp) der Erkrankung und durch den möglichen Nachweis von genetischen Veränderungen in den Knochenmarkzellen bestimmt.
Die Patienten mit einer refraktären Zytopenie im Kindesalter (RCC) werden je nach dem Auftreten bzw. Fehlen von bestimmten Risikofaktoren in unterschiedliche Therapiegruppen eingeteilt (Beobachtung, Stammzelltransplantation oder immunsuppressive Therapie (siehe „Therapie: Wie werden Kinder und Jugendliche mit einem MDS behandelt?“) und haben insgesamt eine gute Prognose mit einer Überlebenswahrscheinlichkeit von 80-90 % in allen Therapiegruppen.
Allerdings haben Patienten mit RCC und einer Monosomie 7 ein sehr hohes Risiko, ein fortgeschrittenes MDS oder eine Leukämie zu entwickeln (Progression). Für diese Patienten wird eine frühzeitige allogene Stammzelltransplantation empfohlen; mit dieser Therapie ist die Prognose gleich gut wie für andere Patienten mit RCC.
Bei einer RCC ist nach erfolgter Stammzelltransplantation das Risiko eines Rückfalls der Erkrankung (Rezidiv) sehr gering. Allerdings stellt jede Stammzelltransplantation eine intensive Therapie dar, bei der das erkrankte Knochenmark nach hochdosierter Chemotherapie durch das gesunde Knochenmark eines Spenders ersetzt wird. Die Patienten sollten daher nach der Stammzelltransplantation lebenslang mindestens einmal im Jahr zur Nachsorgeuntersuchung zu gehen, um Langzeitnebenwirkungen rechtzeitig zu erkennen und behandeln zu können. Auch Patienten, die erfolgreich mit einer immunsuppressiven Therapie (siehe „Behandlung“) behandelt wurden oder die ohne Therapie beobachtet werden, brauchen eine regelmäßige Kontrolle des Blutbildes und eine jährliche Knochenmarkuntersuchung mit einer Knochenmarkstanzbiopsie und einer Zytogenetik, um einen Rückfall oder ein Fortschreiten der Erkrankung frühzeitig erkennen zu können.
Für Kinder und Jugendliche, die an einem fortgeschrittenen MDS (MDS-EB) oder einem sekundären MDS erkrankt sind, gibt es als kurative (heilende) Therapiemöglichkeit nur eine allogene Stammzelltransplantation. Mit dieser Behandlung können ca. 50-60% der Patienten geheilt werden. Dabei haben Patienten mit einer schweren Chromosomenveränderung eine eher ungünstige Prognose.
Literatur
Basisliteratur 
- Niemeyer C, Kratz C: Myelodysplastische Syndrome. in Niemeyer C, Eggert A (Hrsg): Pädiatrische Onkologie und Hämatologie 2017, Springer Verlag; 715 [ISBN: 978-3-662-43685-1]